
is a software package for efficiently sampling trajectories from the chemical and reaction-diffusion master equations (CME/RDME) on high performance computing (HPC) infrastructure using both exact and approximate methods. Along with it, the pyLM problem solving environment allows for customizable simulations.
The current version of Lattice Microbes , v2.3, brings with it the capability to split computations over many GPUs attached to a computer, allowing up to 1 billion grid points on a machine fitted with four current top-of-the-line GPUs. pyLM v1.0 is the initial release.
To install Lattice Microbes v2.3 to build and simulate the HeLa cell model given in:
Zhaleh Ghaemi, Joseph R. Peterson, Martin Gruebele, and Zaida Luthey-Schulten, An in-silico human cell model reveals the influence of organization on RNA splicing, PLOS Computational Biology, 2020, doi:10.1371/journal.pcbi.1007717
Please follow the HeLa Cell Model Installation Guide and the LM Linux Installation Guide. and use LM_2.3_dependency_sources.tar as mentioned in the Installation Guide
Our recent publication provides an overview of the software. In any publication of scientific results based completely or in part on the use of Lattice Microbes and pyLM, please reference:
M. J. Hallock, J. E. Stone, E. Roberts, C. Fry, Z. Luthey-Schulten.
Simulation of reaction diffusion processes over biologically-relevant size and time scales using multi-GPU workstations
Parallel Comput. 40:86-99, 2014, doi: 10.1016/j.parco.2014.03.009
J.R. Peterson, M.J. Hallock, J.A. Cole, and Z. Luthey-Schulten.
A Problem Solving Environment for Stochastic Biological Simulations
PyHPC '13: Proceedings of the 3rd Workshop on Python for High-Performance and Scientific Computing, 2013
Elijah Roberts, John E Stone, and Zaida Luthey-Schulten.
Lattice Microbes: high-performance stochastic simulation method for the reaction-diffusion master equation
J. Comput. Chem., 34(3):245-255, 2013
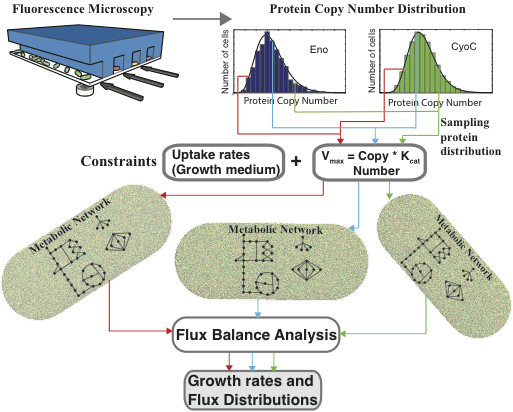
PopulationFBA includes a Matlab script and data file which, when run, samples the copy number distributions of over 350 metabolic proteins in E. coli, and uses them to set constraints on reaction fluxes throughout a comprehensive model of metabolism. A detailed description of this methodology can be found in:
P. Labhsetwar, J.A. Cole, E. Roberts, N.D. Price, Z. Luthey-Schulten. Heterogeneity in protein expression induces metabolic variability in a modeled Escherichia coli population. Proc. Nat. Acad. Sci., 110(34):14006-11, 2013, doi: 10.1073/pnas.1222569110
Includes a compressed directory containing source code for simulating the growth of a colony using the 3DdFBA methodology described in:
Cole JA, Kohler L, Hedhli J, and Luthey-Schulten Z. (2015) Spatially-Resolved Metabolic Cooperativity Within Dense Bacterial Colonies. BMC Syst. Biol. 2015, 9(15), doi:10.1186/s12918-015-0155-1
A method of characterizing allosteric signalling through biomolecular complexes. FEBS Letters, 584(2):376-386, 2010
Alexis Black Pyrkosz, John Eargle, Anurag Sethi, and Zaida Luthey-Schulten
Exit strategies for charged tRNA from GluRS
J. Mol. Biol., 397:1350-1371, 2010
Anurag Sethi, John Eargle, Alexis Black, and Zaida Luthey-Schulten.
Dynamical Networks in tRNA:protein complexes
Proc. Natl. Acad. Sci. U S A, 106(16):6620-6625, 2009
Is a unified bioinformatics analysis environment that allows one to organize, display, and analyze both sequence and structure data for proteins and nucleic acids. Special emphasis is placed on analyzing the data within the framework of evolutionary biology.
In any publication of scientific results based completely or in part on the use of MultiSeq, please reference:
Elijah Roberts, John Eargle, Dan Wright, and Zaida Luthey-Schulten.
MultiSeq: Unifying sequence and structure data for evolutionary analysis.
BMC Bioinformatics, 2006, 7:382.

MultiSeq was primarily developed by Zan Luthey-Schulten, Elijah Roberts, John Eargle, and Dan Wright. We would like to thank Patrick O'Donoghue, Anurag Sethi, John Stone, Michael Bach, and Carl Woese for their help and assistance.
See the MultiSeq press release: http://www.news.uiuc.edu/news/ 06/0918software.html
Is a program that generates a nonredundant set of proteins that represents the topology of the molecular phylogenetic tree of the homologous group.
SeqQR is available on the following platforms:
Representative papers related to SeqQR:
A. Sethi, P. O'Donoghue, and Z. Luthey-Schulten. Evolutionary profiles from the QR factorization of multiple sequence alignments. PNAS, 102:4045, 2005.
P. O'Donoghue and Z. Luthey-Schulten, Evolutionary profiles derived from the QR factorization of multiple structural alignments gives an economy of information. J. Mol. Biol., 346:875, 2005.
P. O'Donoghue, and Z. Luthey-Schulten. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol. Mol. Biol. Rev., 67:550, 2003.
Is an application for the tessellation of 3D volume in and around biological molecules. It creates Tcl script files which are run within VMD to visualize volumes in context with their associated molecular structures.
For help with installation and use of Tessellator, click here.
For supplementary material associated with the paper "Visualizing the Dual Space of Biological Molecules", click here.
Representative paper related to Tessellator:
J Eargle, and Z. Luthey-Schulten, Visualizing the Dual Space of Biological Molecules. Comp. Biol. Chem., 30:219, 2006.
Is an application for the placement atomic ions around 3D biomolecular structures. It uses a pdb file with associated atomic charges to create a grid of electrostatic potential around the structures. An ion is placed at the lowest energy gridpoint, and the grid is then recalculated for the placement of the next ion. Multiple ion types can be placed in a specified order.

Please email support at Lattice Microbes Support for bug reports, installation problems, and help/feature requests. Subscribe to the mailing list for news and announcements at: Lattice Microbes Mailing List
.
Please download both the script (populationFBA_ZLS.m) and data (populationFBA_ZLS_Data.mat) files:
Note: This script requires the use of the COBRA toolbox, which is freely available here: opencobra.sourceforge.net

This download also contains useful MATLAB files for generating input FBA tables and viewing simulation data, as well as a user guide.
Note: This software requires the use of MATLAB and the COBRA toolbox (or Python and cobrapy), which is freely available here:
https://opencobra.github.io
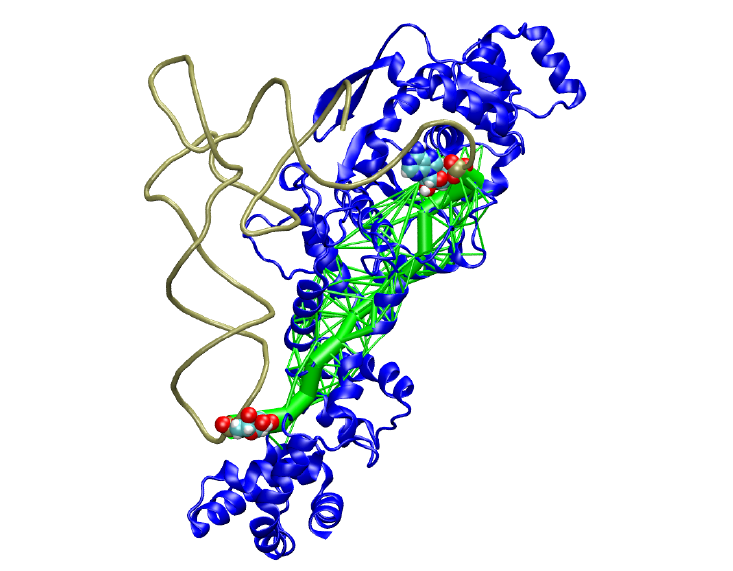
For a general introduction on how to prepare and analyze networks using NetworkView, visit the NetworkView Tutorial page.
An API reference for NetworkView has been generated as Tcldoc documentation.
Please visit our support forums at http://sourceforge.net/projects/latticemicrobes/forums for assistance with installing and using the software.

MultiSeq is included with VMD starting with version 1.8.5.
You can download VMD and MultiSeq from the VMD Download page.
(Note: MultiSeq is located in the Extensions menu as Analysis->MultiSeq)
A paper describing MultiSeq titled "MultiSeq: Unifying sequence and structure data for evolutionary analysis." has been released in BMC Bioinformatics.
Several tutorials are available for learning how to use MultiSeq. Please visit our tutorial page.
The manual provides detailed information on MultiSeq capabilities and options. The manual is available in HTML and PDF.

Combine Sequence and Structure data to gain insight into evolutionary changes in sequence, structure, and function. Compare sequence measures alongside structural measures with just a few mouse clicks.

Quickly and Easily Import Data from many popular files formats or with an integrated BLAST search. Filter search results by sequence identity to eliminate duplicates or by taxonomy to focus a search.
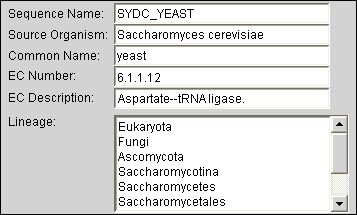
Automatically Find Metadata for sequences and structures from published databases. Use this information (such as taxonomic classification or enzymatic function) during all steps of the analysis process. View and edit metadata via the electronic notebook.

Align Sequences and Structures using two popular alignment algorithms: ClustalW for sequences and STAMP for structures. Perform multiple alignments or profile alignments on entire molecules or just select regions.

Organize Data into custom groupings to ease the process of analyzing large amounts of data. Automatically group by taxonomy to help put the data into an evolutionary context.
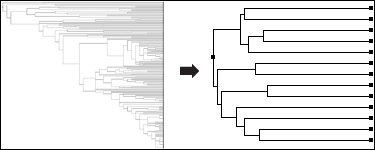
Eliminate Bias and Redundancy by running the QR algorithms for sequence and structure.
View and Manipulate Phylogenetic Trees representing your data by either computing distance trees on the fly or loading precomputed trees from other phylogenetic packages.
Plot Data Measures on a per residue basis with the built-in plotter.
Plus...Work with Nucleic Acid sequences and structures using many of the same techniques as for proteins.Export Data to many popular file formats or create images of alignments, phylogenetic trees, or plots as publication quality PostScript graphics.
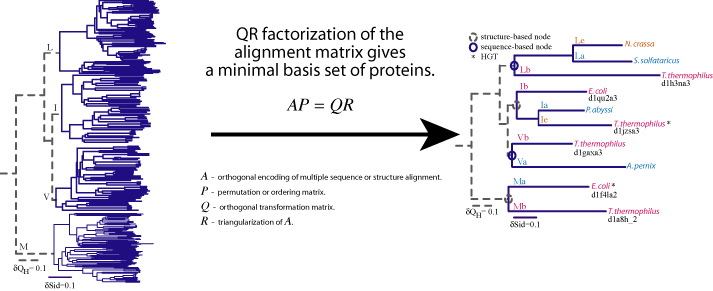
SeqQR is available on the following platforms:
Tessellator is available on the following platforms:
