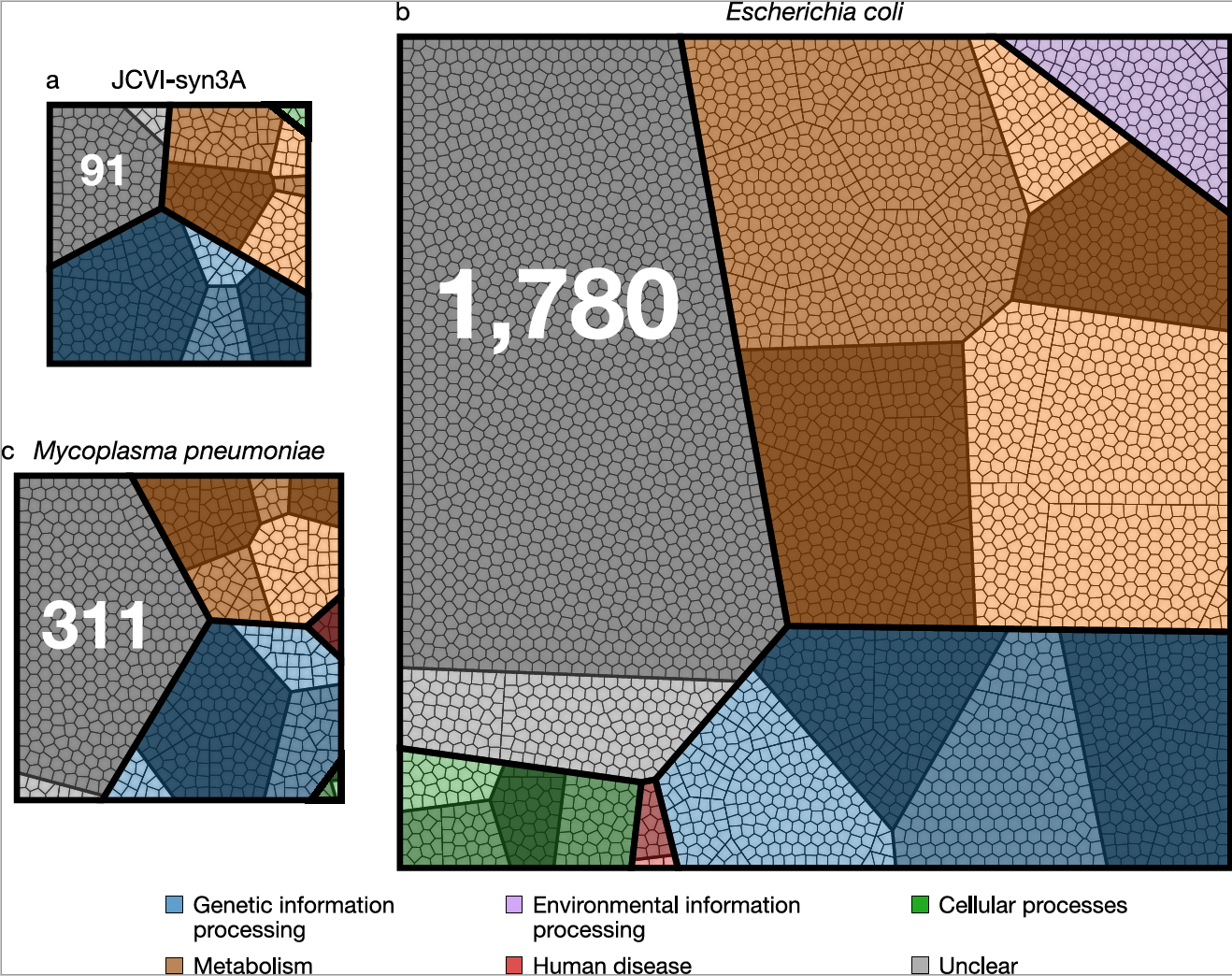
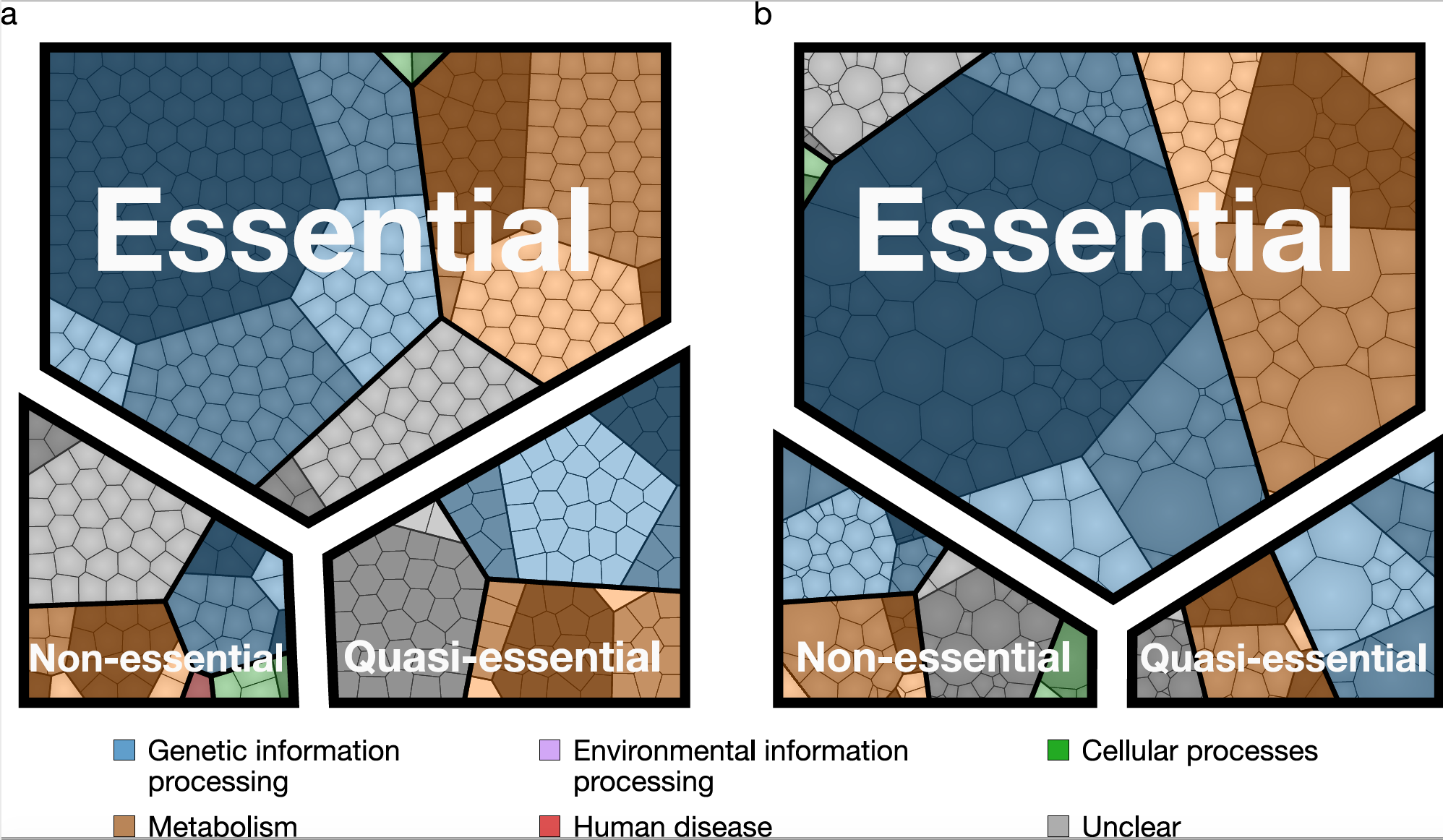
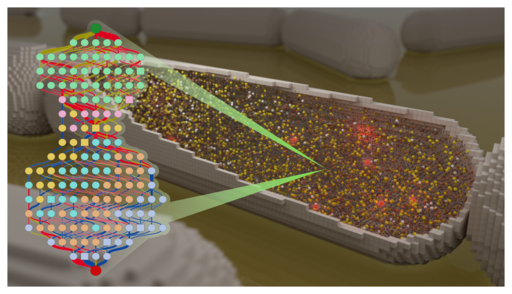
The old question “What is life?” can be recast in genomic terms as “What genes are required for life, and what are their functions?”. We have approached this problem by studying a minimal bacterial cell, JCVI-syn3A. This simple cell is controlled by a genome that was designed based on the genome of the bacterium Mycoplasma mycoides, built by chemical synthesis, and then installed in a living cell. With 493 genes, this genome contains fewer genes than the gene set of any known cell found in nature. Our goal is to build a computer model of the minimal cell based on knowledge of the functions of the individual genes. Such a model tests our understanding of how the cell works. Disagreements between predictions of the model and the observed properties and behavior of the cell help to pinpoint gaps in our knowledge of the gene functions, or the assumptions underlying the model. Our first step was to construct computer model of the network of the essential metabolic reactions in the minimal cell. Our very long term goal -- to make a comprehensive and predictive model of a whole cell, including all of its gene functions. This goal will require that we integrate our metabolic model with the other cellular networks responsible for genetic information processing such as DNA replication, transcription, protein synthesis, and cell division. Using information we collected on the essentiality of all of the genes in the minimal cell including proteomics, metabolomics,and transcriptomics data, we are poised to construct such a complete picture and move us closer to a coarse-grained 3D model. If we can completely understand and model all functions inside an entire minimal cell, then complexifying it should be straightforward. Investigating new functions would have many applications in the field of Synthetic Biology.
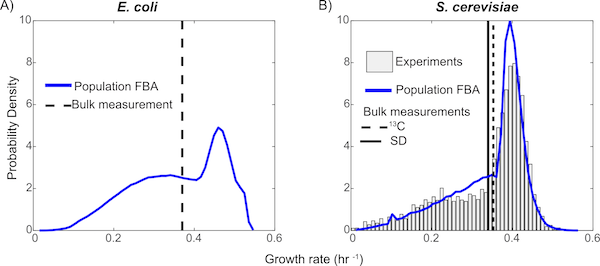
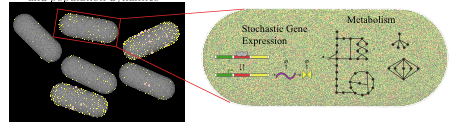
No two living cells are exactly the same. Even cells from a clonal population with identical genomes living in the same environment will express proteins in different numbers simply due to the random nature of the chemistry involved in gene expression. The consequences of this stochastic gene expression are complex and not well understood, especially at the level of large reaction networks like metabolism. Here we investigate how variability in the copy numbers of metabolic enzymes affects how individual cells extract nourishment from their environment and grow. We model 100,000 independent yeast cells, each with their own set of enzyme copy numbers sampled from experimental distributions, and use flux balance analysis (FBA) to compute the optimal way that each cell can use its metabolic pathways---an approach we dubbed Population FBA. We find that enzyme variability gives rise to a wide distribution of growth rates, and several metabolic phenotypes---sub-populations relying on diverse metabolic pathways. Most importantly, we compare the predicted fluxes through the different pathways to experimental values; we find that Population FBA is able to correctly predict Crabtree effect, while traditional FBA, which lacks the proteomics constraints our method imposes, differs both qualitatively and quantitatively from experiment.
Population FBA predicts metabolic phenotypes in yeast. P. Labhsetwar, M.C.R. Melo, J.A. Cole, Z. Luthey-Schulten, PLoS Comput. Biol. 2017, Sep 8, 13(9):e1005728. doi: 10.1371/journal.pcbi.1005728
It's tempting to imagine the living cell as a tiny squishy bag of molecules bouncing around, bumping into each other, and occasionally reacting to form new molecules. It's through these random interactions that cells extract energy from their environment, replicate themselves, and ultimately pass on their genes to the next generation. But importantly, the inherent randomness of this molecular activity also gives rise to significant cell-to-cell variability (or "noise") in things like the numbers of proteins cells produce, and in turn, how the cells behave. Although experiments have shown that most E. coli proteins are dominated by co-called "extrinsic" sources of noise (such as random differences between cells in their RNA polymerase or ribosome concentrations, which make messenger RNA and proteins, respectively), many theoretical models of protein expression routinely overlook extrinsic variability. In a new article, "A Careful Accounting of Extrinsic Noise in Protein Expression Reveals Correlations Among its Sources," appearing in Physical Review E, graduate student John Cole and Professor Zaida Luthey-Schulten present an unprecedentedly complete picture of extrinsic variability in protein expression. Surprisingly, when they compared its predictions to experimental data, they found that it vastly overestimated the variance of most E. coli proteins. This led the researchers to reconsider their assumptions about how different sources of extrinsic noise varied with respect to each other, and ultimately to the realization that several sources had to be correlated in order for the model to match the experiments. The correlations they predict are generally supported by biochemical data, and several can be tested directly in future experiments.
A Careful Accounting of Extrinsic Noise in Protein Expression Reveals Correlations Among its Sources. J.A. Cole, Z. Luthey-Schulten, Phys. Rev. E. 2017, May 12, 95(6):062418. doi: 10.1103/PhysRevE.95.062418
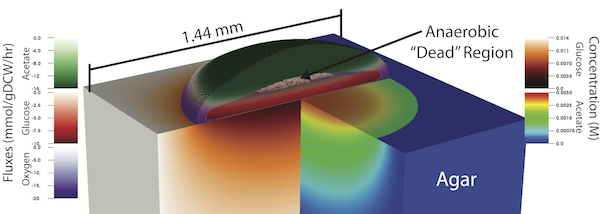
Characterizing the complex spatial and temporal interactions among cells in a biological system (i.e. bacterial colony, microbiome, tissue, etc.) remains a challenge. Metabolic cooperativity in these systems can arise due to the subtle interplay between microenvironmental conditions and the cells's regulatory machinery, often involving cascades of intra- and extracellular signalling molecules. In the simplest of cases, as demonstrated in a recent study of the model organism Escherichia coli, metabolic cross-feeding can arise in monoclonal colonies of bacteria driven merely by spatial heterogeneity in the availability of growth substrates; namely, acetate, glucose and oxygen. Another recent study demonstrated that even closely related E. coli strains evolved different glucose utilization and acetate production capabilities, hinting at the possibility of subtle differences in metabolic cooperativity and the resulting growth behavior of these organisms. Taking a first step towards understanding the complex spatio-temporal interactions within microbial populations, we performed a parametric study of E. coli growth on an agar substrate and probed the dependence of colony behavior on: 1) strain-specific metabolic characteristics, and 2) the geometry of the underlying substrate. To do so, we employed a recently developed multiscale technique named 3D dynamic flux balance analysis which couples reaction-diffusion simulations with iterative steady-state metabolic modeling. Key measures examined include colony growth rate and shape (height vs. width), metabolite production/consumption and concentration profiles, and the emergence of metabolic cooperativity and the fractions of cell phenotypes. Five closely related strains of E. coli, which exhibit large variation in glucose consumption and organic acid production potential, were studied. The onset of metabolic cooperativity was found to vary substantially between these five strains by up to 10 hours and the relative fraction of acetate utilizing cells within the colonies varied by a factor of two. Additionally, growth with six different geometries designed to mimic those that might be found in a laboratory, a microfluidic device, and inside a living organism were considered. Geometries were found to have complex, often nonlinear effects on colony growth and cross-feeding with "hard" features resulting in larger effect than "soft" features. These results demonstrate that strain-specific features and spatial constraints imposed by the growth substrate can have significant effects even for microbial populations as simple as isogenic E. coli colonies.
Parametric Studies of Metabolic Cooperativity in Escherichia coli Colonies: Strain and Geometric Confinement Effects.
J.R. Peterson, J.A. Cole, Z. Luthey-Schulten, PLoS ONE, 2017, Aug 18, 12(8):e0182570. doi: 10.1371/journal.pone.0182570
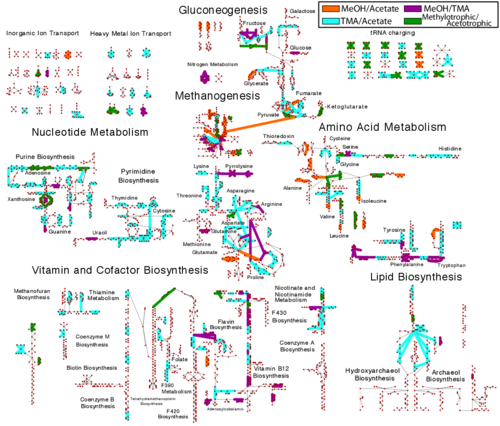
While a few studies on the variations in mRNA expression and half-lives measured under different growth conditions have been used to predict patterns of regulation in bacterial organisms, the extent to which this information can also play a role in defining metabolic phenotypes has yet to be examined systematically. Here we present the first comprehensive study for a model methanogen. We use expression and half-life data for the methanogen Methanosarcina acetivorans growing on fast- and slow-growth substrates to examine the regulation of its genes. Unlike Escherichia coli where only small shifts in half-lives were observed, we found that most mRNA have significantly longer half-lives for slow growth on acetate compared to fast growth on methanol or trimethylamine. Interestingly, half-life shifts are not uniform across functional classes of enzymes, suggesting the existence of a selective stabilization mechanism for mRNAs. Using the transcriptomics data we determined whether transcription or degradation rate controls the change in transcript abundance. Degradation was found to control abundance for about half of the metabolic genes underscoring its role in regulating metabolism. Genes involved in half of the metabolic reactions were found to be differentially expressed among the substrates suggesting the existence of drastically different metabolic phenotypes that extend beyond just the methanogenesis pathways. By integrating expression data with an updated metabolic model of the organism (iST807) significant differences in pathway flux and production of metabolites were predicted for the three growth substrates. This study provides the first global picture of differential expression and half-lives for a class II methanogen, as well as provides the first evidence in a single organism that drastic genome-wide shifts in RNA half-lives can be modulated by growth substrate. We determined which genes in each metabolic pathway control the flux and classified them as regulated by transcription (e.g. transcription factor) or degradation (e.g. post-transcriptional modification). We found that more than half of genes in metabolism were controlled by degradation. Our results suggest that M. acetivorans employs extensive post-transcriptional regulation to optimize key metabolic steps, and more generally that degradation could play a much greater role in optimizing an organism's metabolism than previously thought.
Genome-wide gene expression and RNA half-life measurements allow predictions of regulation and metabolic behavior in Methanosarcina acetivorans.
J.R. Peterson, S. Thor, L. Kohler, P.R.A. Kohler, W.W. Metcalf, Z. Luthey-Schulten, BMC Genomics, 2016, Nov 16, 17(1):924 doi: 10.1186/s12864-016-3219-8
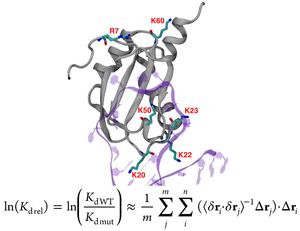
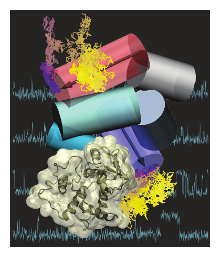
Protein-RNA complexes are increasingly important in our understanding of cell signaling, metabolism, and transcription. Electrostatic interactions play dominant role in stabilizing such complexes. Using conventional computational approaches, very long simulations of both bound and unbound states are required to obtain accurate estimates of complex dissociation constants (Kd). Here, we derive a simple formula that offers an alternative approach based on the theory of fluctuations. Our method extracts a strong correlate to experimental Kd values using short molecular dynamics simulations of the bound complex only. To test our method, we compared the computed relative Kd values to our experimentally measured values for the U1A-Stem Loop 2 (SL2) RNA complex, which is one of the most-studied protein-RNA complexes. Additionally we also included several experimental values from the literature, to enlarge the data set. We obtain a correlation of r = 0.93 between theoretical and measured estimates of Kd values of the mutated U1A protein-RNA complexes relative to the wild type dissociation constant. The proposed method increases the efficiency of relative Kd values estimation for multiple protein mutants, allowing its applicability to protein engineering projects.
Role of Electrostatics in Protein-RNA binding: The Global vs. the Local Energy Landscape. Z. Ghaemi, I. Guzman, D. Gnutt, Z. Luthey-Schulten, M. Gruebele, J. Phys. Chem. B, 2017, Aug 14. doi: 10.1021/acs.jpcb.7b04318
Estimation of Relative Protein-RNA Binding Strengths from Fluctuations in the Bound State. Z. Ghaemi, I. Guzman, J.J. Baek, M. Gruebele, Z. Luthey-Schulten. J. Chem. Theory Comput., 2016, Aug 16. doi:10.1021/acs.jctc.6b00418
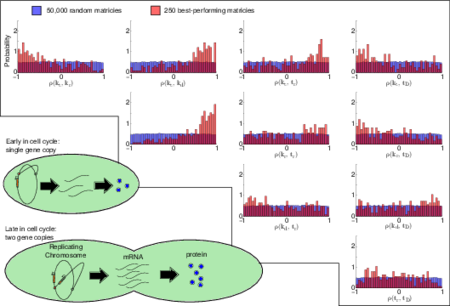
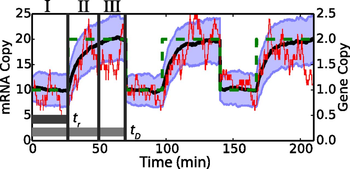
There are several sources of fluctuations in gene expression. Here we study the effects of time-dependent DNA replication, itself a tightly controlled process, on noise in mRNA levels. Stochastic simulations of constitutive and regulated gene expression are used to analyze the time-averaged mean and variation in each case. The simulations demonstrate that to capture mRNA distributions correctly, chromosome replication must be realistically modeled. Slow relaxation of mRNA from the low copy number steady state before gene replication to the high steady state after replication is set by the transcript’s half-life and contributes significantly to the shape of the mRNA distribution. Consequently both the intrinsic kinetics and the gene location play an important role in accounting for the mRNA average and variance. Exact analytic expressions for moments of the mRNA distributions that depend on the DNA copy number, gene location, cell doubling time, and the rates of transcription and degradation are derived for the case of constitutive expression and subsequently extended to provide approximate corrections for regulated expression and RNA polymerase variability. Comparisons of the simulated models and analytical expressions to experimentally measured mRNA distributions show that they better capture the physics of the system than previous theories.
Effects of DNA Replication on mRNA Noise. J.R. Peterson*, J.A. Cole*, J. Fei, T.J. Ha and Z. Luthey-Schulten, Proc. Nat. Acad. Sci., 2015, Nov 11, 112(52): 15886-15891 doi:10.1073/pnas.1516246112
Translation is the universal process that synthesizes proteins in all living cells. Central to translation is the ribosome, which itself constitutes approximately one fourth of a bacterial cell's dry mass. Biogenesis of the ribosome, together with all cellular activities involved in translation consume a significant budget of the cell's total energy. The ribosome consists of two subunits, each constructed of bare ribosomal RNA and a large number of ribosomal protein. These protein bind to the nascent ribosomal subunits in an hierarchy where the binding of a protein is dependent on the prior binding of an other. Here we describe a model of the biogenesis of the 30S ribosomal small unit based on kinetic data from bulk experiments as well as single molecule experiments and molecular dynamics simulation. By using the thermodynamic dependencies of r-protein we construct a chemical reaction network enumerating all 1600 possible intermediates and all 7000 binding reactions. The full network is then reduced iteratively by removing the least utilized intermediate until an error tolerance of 1% RMS is reached. The reduced network is able to confirm the known 5' to 3' binding order as well as predict all important intermediates. The reduced network shows a single pathway constructing the 5' domain, followed by two independent pathways constructing the central and 3' domain, completed by a single pathway binding the remaining protein. This reduced network will be used to construct whole-cell models of ribosome biogenesis allowing detailed predictions of spatial distributions of ribosomes
Ribosome biogenesis in replicating cells: integration of experiment and theory. T.M. Earnest, J.A. Cole, J.R. Peterson, T.E. Kuhlman, Z. Luthey-Schulten,Biopolymers, 2016, Jul 22, 105(10): 735-751 doi:10.1002/bip.22892
Towards a whole-cell model of ribosome biogenesis: Kinetic modeling of SSU assembly. T.M. Earnest, J. Lai, K. Chen, M.J. Hallock, J.R. Williamson, Z.A. Luthey-Schulten, Biophysical Journal. 2015, Volume 109, Number 6, pages 1117-11135. doi: 10.1016/j.bpj.2015.07.030
Due to the stochastic nature of gene expression, genetically identical cells inhabiting the same environment can vary significantly in their numbers of key enzymes, which in turn results in strikingly different cellular behaviors. This cell-to-cell variability can manifest itself through differences in growth rates, usage of specific biochemical pathways, and the types of metabolic byproducts produced by each cell. Incorporating data from studies of gene regulation and protein distributions in single cells, we developed a population flux balance methodology that identified several behavioral subtypes within a population grown on minimal medium. Our computer model predicts the emission of acetate for slow growing subpopulations and pathway selection to balance energy (glycolysis pathway) and protein costs (ED pathway) as a function of growth. The research also suggests that tracking the behavior of a few genes "may be sufficient to capture most of the metabolic variability of the entire population. Our investigations provide the first calculations linking variation in specific pathway usages to the growth rate distribution of a microbial population. By looking beyond the average growth rate of a colony, our work provides insight into the different strategies used by bacteria for survival and is an important step in the development of physical systems biology.
Heterogeneity in protein expression induces metabolic variability in a modeled Escherichia coli population. Labhsetwar P, Cole JA, Roberts E, Price ND, Luthey-Schulten ZA, Proc Natl Acad Sci U S A. 2013, Volume 110, pages 14006-11. doi: 10.1073/pnas.1222569110
Spatially resolved stochastic simulation is a valuable technique for studying reactions on time and size scales relevant for biological systems. Here, CPLC faculty ZLS and postdoc ER introduce the Lattice Microbes GPU-based software package for simulating complex biochemical reaction networks on the level of the whole cell. Our approach integrates data from cyroelectron tomography, proteomics, and single molecule spectroscopy to capture the molecular crowding within cells. The software performs either well-stirred or spatially resolved stochastic simulations with approximated cytoplasmic crowding in a fast and efficient manner. Our new algorithm efficiently samples the reaction-diffusion master equation using NVIDIA graphics processing units and is shown to be two orders of magnitude faster than exact sampling for large systems while maintaining an accuracy of ∼0.1%. Display of cell models and animation of reaction trajectories involving millions of molecules is facilitated using a plug-in to the popular VMD visualization platform.
Lattice microbes: High-performance stochastic simulation method for the reaction-diffusion master equation. Elijah Roberts, John E. Stone and Zaida Luthey-Schulten, Journal of Computational Chemistry, Volume 34, 2013, Pages: 245-255. doi: 10.1002/jcc.23130
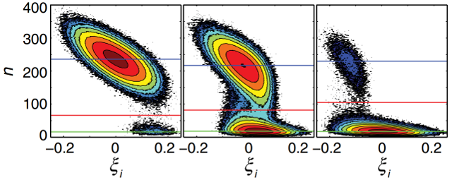
In a new collaborative direction between Luthey-Schulten and Goldenfeld, the work is primarily analytical, and will help us understand the emergence of heterogenous cell populations in initially clonal bacterial populations. Biological systems are of great interest to statistical physicists, because they contain a large number of strongly fluctuating degrees of freedom, but not enough that they are in the thermodynamic limit. Accordingly the noise characteristics and the dynamical behavior of such systems pose a unique challenge to theory that is rarely encountered in other areas of physics, leading to important biological phenomena. A population of clonal cells can become phenotypically differentiated as a result of environmental (i.e. extrinsic) noise and intrinsic noise, such as number fluctuations. These phenomena are now well-understood in the case where intrinsic gene expression stochasticity is the key noise source. But what is the role of extrinsic noise, arising from cell-to-cell variations in (e.g.) ribosome or RNA polymerase number, thus equally affecting each gene within the cell? How do mean switching times between allowed states depend on the extrinsic noise?
To address this, Goldenfeld and Luthey-Schulten have solved the problem of phenotype switching due to a single self-regulating gene with positive feedback. The technical advance that they and their CPLC associated postdocs introduced in this context was to use WKB and Hamilton-Jacobi methods to go beyond simple mass-action results. The key result was that the different phenotypes' lifetime is significantly altered, with increased parameter range for bistability. The mean switching time is lowered by many orders of magnitude even for a very moderate amount of extrinsic noise, which is important for bacterial communities that exploit heterogeneity in order to inhabit new ecological niches, for example. The semi-analytical results were validated through state of the art stochastic simulations available through the Luthey-Schulten’s Lattice Microbes to go beyond simple mass-action results.
Extrinsic Noise Driven Phenotype Switching in a Self-Regulating Gene. Michael Assaf, Elijah Roberts, Zaida Luthey-Schulten, and Nigel Goldenfeld. Physical Review Letters. Vol. 111, 2013, Pages: 058102- doi: 10.1103/PhysRevLett.111.05810
To our knowledge, this collaboration between CPLC faculty Ha and Luthey-Schulten with Woodson at Johns Hopkins presents the first use of single molecule spectroscopy and all-atom simulations to obtain the frequency and direction of helix motions (not only populations) in a large RNA-protein complex. The first proteins to bind the RNA change the rRNA structure in a way that makes it easier for later proteins to join. How ribosomal proteins recognize the RNA and change its structure so that other proteins can bind is not known. To address this gap, we used two and three-color single molecule FRET and molecular dynamics simulations to observe encounters between E. coli protein S4 and the 5’ domain of 16S rRNA in real time with precision and clarity unprecedented for any RNA-protein complexes. S4 is one of the first proteins to bind the 16S rRNA, and is needed to nucleate 30S ribosome assembly and for the fidelity of translation. At first, the S4-rRNA complex is dynamic and samples many structures, but after a few seconds, it converts to stable complexes that flip between two structures, a native structure and a non-native intermediate. Unexpectedly, nearly all successful S4 binding trajectories pass through the non-native intermediate, contradicting the usual assumption that proteins prefer to bind the natively folded RNA.
Protein-guided RNA dynamics during early ribosome assembly. Hajin Kim, Sanjaya C. Abeysirigunawardena, Ke Chen, Megan Mayerle, Kaushik Ragunathan,Zaida Luthey-Schulten, Taekjip Ha and Sarah A. Woodson. Nature. Vol. 506, 2014, Pages: 334-338 doi: 10.1038/nature13039.
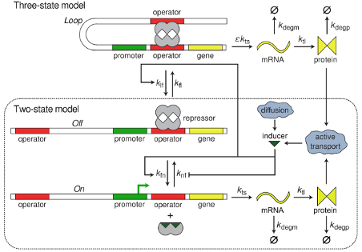
We develop a model of the lac genetic switch that describes the switching and coexistence of phenotypes as a function of extracellular lactose concentration. We present a first stochastic treatment of a gene–mRNA–protein model of the lac operon in E. coli interacting with extracellular inducer that includes transitions to looped DNA states. This highly interdisciplinary project, involving 2 post-docs and one graduate student, uses tools from physics and new mathematical tools to explain statistical aspects of stochastic gene expression.
DNA looping increases the range of bistability in a stochastic model of the lac genetic switch. Tyler M Earnest, Elijah Roberts, Michael Assaf, Karin Dahmen and Zaida Luthey-Schulten. Physical Biology. Vol. 10, No. 2, 2013, Pages: 026002- doi: 10.1088/1478-3975/10/2/026002